Abstract
Cystic fibrosis and pulmonary biofilms
Kenneth Nugent MD
Corresponding author: Kenneth Nugent
Contact Information: Kenneth.Nugent@ttuhsc.edu
DOI: 10.12746/swrccc.v11i49.1233
ABSTRACT
Cystic fibrosis (CF) is an autosomal recessive disorder that involves the cystic fibrosis transmembrane conductance regulator (CFTR). This protein is an anion channel that transfers chloride and bicarbonate from an intracellular location to an extracellular location. This transfer supports the formation of a normal periciliary fluid layer that is essential for ciliary function and the clearance of particulates from bronchi. Changes in mucociliary function can result in chronic bronchitis and recurrent infections. Studies using micro computed tomography of explanted lungs from CF patients have demonstrated that there is a significant reduction in the number of terminal bronchi and that approximately 50% of these bronchi are obstructed with secretions. These airways become colonized with bacteria, such as Pseudomonas aeruginosa, which can form biofilms, and this results in chronic inflammation and chronic airway damage. Biofilm formation can be predicted when mucoid colonies of pseudomonas grow out on routine bacterial cultures of sputum. Studies on the prognosis of patients with CF have demonstrated that there is lack of clear genotype-phenotype correlation, but one recent longitudinal study did report an association between more severe CFTR genotypes and clinical outcomes. In addition, the degree of pulmonary dysfunction and pseudomonas colonization, especially with drug resistant pseudomonas, predict poor outcomes. Bacteria in biofilms are more resistant to antibiotics, and the structure of biofilms limits host defense responses. Treatment approaches include the use of more than one antibiotic, the addition of inhaled antibiotics, the use of enzymes that degrade biofilms, and the use of small molecules that inhibit community metabolism in biofilms.
Keywords: cystic fibrosis transmembrane conductance regulator, extracellular secretions, periciliary fluid, Pseudomonas aeruginosa, biofilms
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive disorder that involves the cystic fibrosis transmembrane conductance regulator (CFTR), an anion channel in the luminal membrane of epithelial cells that regulates the volume and composition of exocrine secretions.1 The disease presentation in CF usually involves the lungs, but other organs, including the pancreas, liver, intestine, and reproductive tract, can be involved. The CFTR is a membrane protein that functions as a passive conduit for chloride and bicarbonate transfer across the plasma membrane. This transfer is supported by the hydrolysis of ATP. Water also moves out of the cell onto epithelial surfaces through other channels in association with the chloride and bicarbonate transfer, and this results in a normal periciliary fluid layer and normal ciliary function.
The most common mutation is the F508 deletion. This results from a single omission of phenylalanine at position 508, which leads to a folding abnormality. Protein maturation is stopped in the endoplasmic reticulum and the protein fails to reach the plasma membrane. It then undergoes degradation in the cytoplasm. This is a class II defect and accounts for 70% of defective alleles in the United States (Figure 1).1 Other mutations result in the lack of protein synthesis (Class I), reduce the synthesis of the protein (class V), reduce channel function of the protein (classes III and IV), or lead to accelerated turnover of the protein (class VI). The disease occurs in approximately 1 out of 3300 live births in the United States.
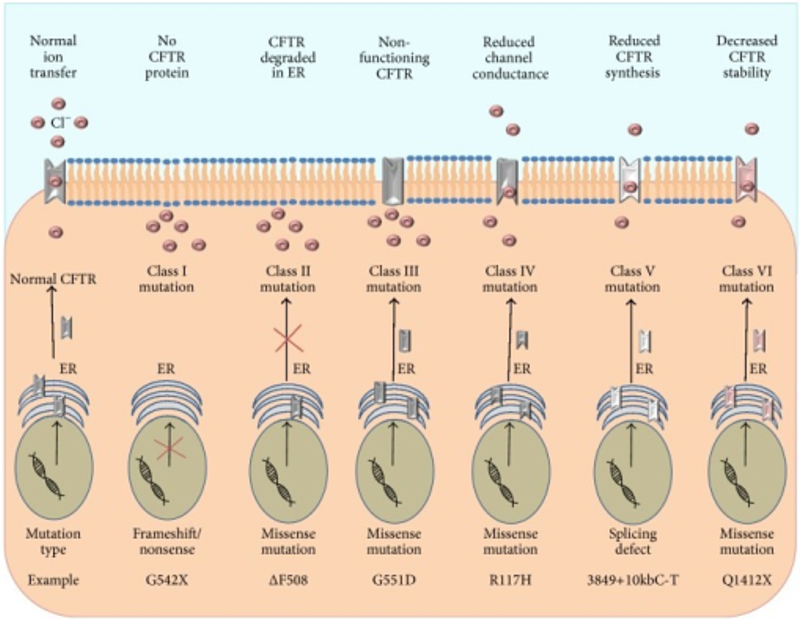
Figure 1. Classification of CFTR mutations. In healthy CFTR sufficient cells, the functional CFTR protein is correctly trafficked to the plasma membrane. Class I mutations result in a lack of CFTR protein synthesis. Class II mutations block CFTR processing, where misfolded protein is degraded in the ER. Class III mutations affect the regulation of the CFTR, where the CFTR channel is less functional. Class IV mutations alter the CFTR conductance of Cl−. Class V mutations lead to reduced synthesis of functional CFTR. Class VI mutations result in accelerated turnover of CFTR protein on the cell surface. Lavelle GM, et al. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. Biomed Res Int. 2016; 2016:5258727. Accessed through OPENi database in the TTUHSC library on 9-10-2023.
PATHOLOGICAL CHANGES IN CF LUNGS
Computed tomography (CT) of the lung provides important information about the type and extent of disease in patients with CF. The CT images in Figure 2 illustrate common changes, including bronchiectasis, airway occlusion with secretions, and thickened bronchial walls.
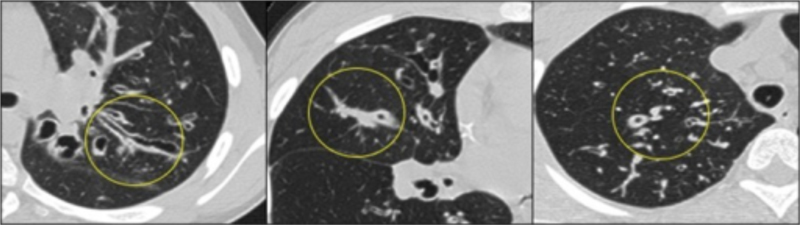
Figure 2. Tepper LA, et al. The development of bronchiectasis on chest computed tomography in children with cystic fibrosis: can pre-stages be identified? Eur Radiol. 2016 Dec;26(12):4563–4569. Accessed through the OPENi database at the TTUHSC library on 9-10-2023.
The pathologic changes in patients with CF have also been studied using explanted lungs obtained at the time of lung transplantation, using both standard gross and microscopic pathologic techniques and CT studies of sections of the lung parenchyma. Boon and co-investigators used multi-detector CT and micro CT on 11 air-inflated CF expanded lungs.2 With multi-detector CT examinations, the number of visible airway bifurcations in CF lungs was increased compared to control lungs, and there was a significant increase in airway diameter per airway generation. The number of visible airways increased from airway generation 9 onward; the diameter increased from generation 6 onward. Thirty-five percent of the airways were obstructed starting with airway generation 6. The median number of visible terminal bronchi per milliliter lung tissue was significantly reduced in CF core lung samples, and the median diameters and cross-sectional areas were also reduced. In CF lungs approximately 50% of the terminal bronchi remained open over their entire course; approximately 50% of the obstructive bronchi were completely obstructed, and 50% reopened downstream from the obstruction. These CF lungs had an increased percent tissue, consistent with inflammation in lung tissue. This study demonstrated that there are important changes in the bronchi in CF lungs that increase the likelihood of infection and alter gas exchange through the changes in ventilation-perfusion ratios.
Lammertyn et al. studied the types of inflammatory cells in explanted CF lungs. This study included 20 lungs collected at the time of transplantation with comparisons to 20 control lung tissues.3 These lungs were sectioned and stains for neutrophils, eosinophils, mast cells, dendritic cells, macrophages, T cells, and B cells. These investigators found that all inflammatory cell types, except for B cells, were significantly increased in CF lungs. There was a disproportionate increase of neutrophils around the airways in patients with CF. In addition, they found increased numbers of lymphoid follicles around airways, in lung parenchyma, and in perivascular regions. These lymphoid aggregates had increased numbers of CD4 and CD8 T cells. This study indicates there is a complex inflammatory process in patients with end-stage CF. There are increased numbers of cells involved in both innate immunity and adaptive immunity, and adaptive immune responses may contribute to the pathogenesis of CF lung disease.
MICROBIAL COMMUNITIES IN CF LUNGS
Studies with explanted lungs provide the clinical material needed to analyze associations between bacterial populations in different lung regions and the association between the presence of bacteria and lung damage. Einarsson et al. analyzed the microbial community composition of explanted CF lungs in patients who were undergoing lung transplantation.4 Lung tissue and luminal secretions were cultured and analyzed using DNA sequencing; in addition, lung tissue was studied using micro-CT scoring. There was a significant intra-and inter-patient variability. The samples from the same patient were in general more similar to each other than samples from different patients. There was significant dissimilarity between CF lung tissue and donor lung tissue. The dissimilarity between different patients was higher than dissimilarity between tissue specimens in the same patient. Microbial diversity was significantly lower in CF tissue and microbial dominance was significantly higher. The relative abundance of Pseudomonas, Achromobacter, Staphylococcus, Stenotrophomonas, Provotella, and Veillonella species differed significantly in CF patients, and CF tissue had increased numbers or abundance of Pseudomonas, Staphylococcus, and Achromobacter species. Donor tissue had a relative abundance of Streptococcus species. There was no significant relationship between the microbial community and location in the upper, middle, and lower lobes and lung damage based on CT scores.
The microbial population differed between patients with CF. There was also a difference at times between the microbial population in luminal secretions and in the lung tissue; this suggests that sputum cultures may not provide an accurate representation of bacteria in the lung parenchyma. In addition, there was no obvious difference between regional microbial variation and structural lung damage. However, a single one time sample of the lung at the time of the explantation probably does not provide a satisfactory understanding of the time course and complexity of chronic infections in this chronic disease.
Malhotra and colleagues analyzed the distribution of Pseudomonas aeruginosa and its association with regional inflammation in patients with CF.5 They used bronchoalveolar lavage in 14 patients to collect fluid for culture and cytokine analysis from all 6 lobes of the lung. They demonstrated that pseudomonas mucoid and nonmucoid variants were distributed homogeneously throughout the lung. However, infections in regions with mucoid variants had significantly higher levels of regional inflammation based on assays of IL-1, TNF, IL-6, and IL-8. Therefore, this study indicates that the presence of mucoid pseudomonas had a strong effect on or an association with ongoing inflammation.
PSEUDOMONAS AERUGINOSA
Pseudomonas is a nonfastidious, motile, Gram-negative rod that grows on most common laboratory media. On laboratory media, it synthesizes a pigment that is yellow to dark green (Figure 3). Some colonies have a mucoid appearance due to the production of large amounts of exopolysaccharides or alginate. Pseudomonas has multiple potential virulence factors, including lipopolysaccharide, proteases, phospholipases, pyocyanin, and rhamnolipids, a class of glycolipids.6 The bacteria in chronic pseudomonas infections have changes in phenotype that leads to less virulence. Injury during chronic infection likely develops secondary to pseudomonas virulence factors and an attenuated host defense response that is present for long periods of time, possibly years.
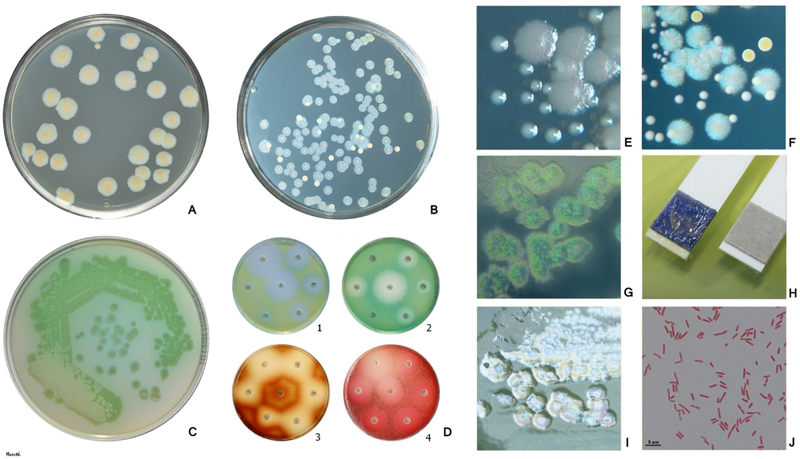
Figure 3. Pseudomonas aeruginosa is a nonfastidious, Gram-negative (Fig. J), oxidase positive (Fig. H), nonfermenting rod. P. aeruginosa produces pyoverdin, a water-soluble yellow-green pigment (Fig. A, C, D1, D2). Many P. aeruginosa strains also produce the blue pigment pyocyanin (Fig. B, F). When pyoverdin combines with pyocyanin, the bright green color characteristic of P. aeruginosa is created (Fig. C). Some strains rarely produce other pigments: brown pyomelanin (D3) or red pyorubrin (D4). For the isolation and presumptive identification from clinical and environmental samples a selective agar containing cetrimide can be used (Fig. G). Fig. A, B Trypticase soy agar (B: P. aeruginosa + S. aureus), Fig. C, D, I Mueller-Hinton agar Fig. E Larger colonies of P. aeruginosa and smaller colonies of Enterococcus faecalis on tripticase soy agar. Reflected light. Fig. F Bluish colonies of P. aeruginosa, yellow colonies of S. aureus and white colonies of Enterococcus faecalis on trypticase soy agar. Fig. I Plaques are frequently found in freshly isolated strains, especially in the area of confluent growth. In some strains these are caused by phage, in others by bacteriocins. Accessed through a Wikimedia Commons on 9-10-2023.
BIOFILMS AND HOST DEFENSES
Biofilms are structured communities of sessile bacteria encapsulated in an extracellular polymeric substance matrix (Figure 4).7 This structure provides homeostasis and stability in a fluctuating and potentially harsh environmental conditions for the bacteria. In particular, biofilms can protect bacteria from host defenses and antibiotics. The matrix is composed of polysaccharides, proteins, extracellular DNA, and lipids. The formation and dispersal of biofilms are regulated by quorum sensing molecules, di-cyclic guanosine monophosphate, and micro RNAs. The two quorum sensing pathways in pseudomonas include two acyl-homoserine lactone signaling molecules.
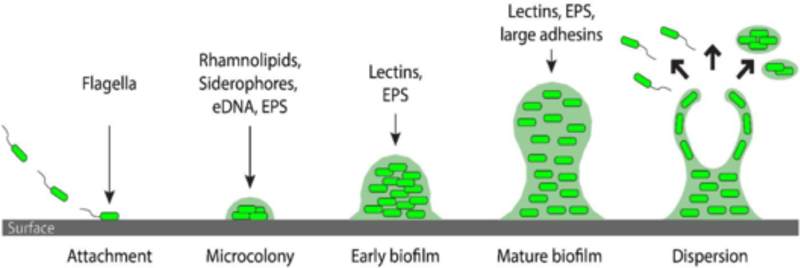
Figure 4. The first stage of biofilm formation is the attachment of bacteria to a surface, which has been associated with flagella. In the second stage, the ability to produce rhamnolipids (swarming), siderophores (iron availability), eDNA and EPS (matrix formation) are thought to be essential for microcolony formation. In the subsequent stages of biofilm maturation, lectins, adhesins and EPS are important for the proper building of the matrix and localization of its components. The final stage is biofilm dispersion. Accessed from Wikimedia on 9-10-2023.
The epithelium provides the initial host defense against pseudomonas infections. Biofilm formation on these surfaces inhibit the phagocytic and killing activity of both neutrophils and macrophages. In addition, pseudomonas can inactivate the complement system by cleavage of complement components. Pseudomonas isolated from CF patients commonly have inactivation of the MucA gene, which results in increased production of the extracellular polysaccharide alginate. This gene inactivation also promotes a stress response that represses bacterial metabolism, motility, and virulence. The decreased metabolic activity of bacteria in biofilms may reduce their inhibition by antibiotics. Bacterial resistance to antibiotics also involves drug inactivation, alterations in drug binding sites, increased drug efflux, and the use of alternative metabolic pathways to bypass drug effects. In addition, antibiotics may not be able to penetrate into biofilms. Approaches to bacterial control or elimination in the biofilms include quorum sensing inhibition, quorum quenching, augmented host defense response, inhibition of cyclic di- GMP signaling, and the use of antimicrobial peptides.
Pseudomonas aeruginosa was the first biofilm infection described in humans.8,9 However, other bacterial pathogens associated with CF can also form biofilms. The paranasal sinuses often become colonized before the lower airways. Pseudomonas is then aspirated into the lungs during episodes of viral infection. These episodes of aspiration and colonization eventually lead to chronic lung infection. Neutrophils are found in luminal secretions in both respiratory bronchi and conductive bronchi. This can lead to tissue damage and loss of lung function. Chronic pseudomonas infection leads to IgG antibody responses against pseudomonas components, including alginate. Several diagnostic methods can identify biofilms. Microscopy can identify aggregates of bacteria which are 4 to 100 µm in size in a matrix dominated by alginate which can be stained by Alcian blue. The bacteria in biofilms form mucoid colonies when grown on agar. There are IgG antibody responses in the serum to pseudomonas antigens. Chronic pseudomonas infection in the paranasal sinuses is also characterized by biofilm formation, and there is secretory IgA against alginate. The presence of alginate into sputum also provides diagnostic information about biofilms.10,11
Biofilms are complex structures that can be characterized by physical traits, such as size, and matrix, and by phenotypic traits, such as metabolic activity and antibiotic tolerance. Experimental studies using in vitro methods provide the best approach to study these structures. However, it is likely that biofilms in patients with infections are far more complicated, and their structure and metabolism are affected by nutrient availability, host defense responses, and intermittent antibiotic use.12
PROGNOSIS
The importance of chronic bronchial infection becomes apparent in longitudinal studies. Durda-Masny et al. prospectively followed 124 patients with CF. Patients were recruited in 2010 and followed annually for the next 9 years.13 At the start of the study, ages ranged from 18–41. Patients undergoing lung transplantation, patients who were pregnant, patients who smoke, and patients who used glucocorticoids were excluded from the study. Important characteristics included the type of mutation, nutritional status, lung function, and pseudomonas prevalence in sputum cultures. All four factors affected survival. The shortest life expectancy was observed in patients with a severe type mutation on both alleles, in patients with a FEV1 percent predicted less than 40%, in patients with extensively drug-resistant or pan drug-resistant pseudomonas in sputum cultures, and in patients whose BMIs were below 18.5. Logistic regression analysis indicated that the risk of death doubled (OR = 2.06; 1.29, 2.28) when pseudomonas was present in sputum and went up 8-fold (OR = 8.11; 1.67, 38.15) when the bacteria had acquired antibiotic resistance. A summary of the odds ratio for increased mortality during follow up included: FEV1% below normal (OR = 5.83; 2.51, 13.52), BMI below normal (OR = 4.17; 1.65, 10.5), severe CFTR mutation (OR = 2.35; 1.17, 4.71), pseudomonas present (OR = 2.06; 1.29, 2.28), and pseudomonas antibiotic resistance (OR = 8.11; 1.67, 38.15). While there is lack of clear genotype-phenotype correlation, this study does report an association between more severe CFTR genotypes and clinical outcome.
STRATEGIES FOR CONTROLLING BACTERIAL BIOFILMS
The best strategy for managing and preventing bacterial colonization and infection in the CF lung is to prevent biofilm formation. These CFTR modulators have the potential to do this or at least to slow the process. There is extensive literature on CFTR modulators, but these important drugs are not discussed in this review.1
After biofilms are formed, management becomes more difficult.14 The minimal antibiotic concentration needed for bactericidal activity may be 100 to 1000-fold higher against biofilm-based bacteria than planktonic or free-floating bacteria. Some antibiotics target only growing bacteria and therefore have limited effect on bacteria with low metabolic activity, so-called persister cell, in the lung. Some antibiotics are unable to penetrate biofilms. Antibiotic combinations may be more effective than single drugs alone and liposomal preparations of antibiotics may have a better entry into biofilm. Also, bacteriophages can directly kill bacteria. They can be used to target particular bacterial species, and they can lyse metabolically dormant bacteria.
Some therapeutic approaches target the biofilm matrix and try to break down the matrix.15 For example, alginate lysase can break down alginate, which forms a sticky matrix of the biofilm, and some oligomers of alginate can increase the pore size in the matrix and improve diffusion into the matrix. Some enzymes can degrade exopolysaccharides, and deoxyribonuclease can degrade extracellular DNA. Another target is intracellular signaling pathways. Nitric oxide increases the concentration of specific phosphodiesterase enzymes which then reduce levels of cyclic di-GMP. This leads to the dispersal of bacteria from the biofilm, and these planktonic bacteria are more susceptible to antibiotics. Azithromycin can disrupt quorum sensing, and gallium can disrupt iron metabolism which is essential for bacterial growth.
SUMMARY
The CFTR ion channel transfers chloride across cellular membranes to maintain the composition of extracellular secretions. This function is essential in the lung to maintain an optimal pericellular fluid base for ciliary function to transfer a particulate material, including bacteria, out of the lung. Patients with cystic fibrosis have abnormal bronchi with increased secretions, which result in recurrent infections. Pseudomonas colonizes these regions of the lung and creates biofilms. These biofilms increase the survival of bacteria, reduce antibiotic effects on the bacteria, and limit the recruitment of neutrophils into these regions. The presence of mucoid pseudomonas on simple bacterial cultures of respiratory secretions indicates the presence of biofilms. The treatment of infections in these patients might have better outcomes if more than one antibiotic is used, if inhaled antibiotics are added to the treatment regimen, and if chemicals that can break down biofilms are also used. The best treatment results are likely produced by CFTR modulators that maintain normal anion channel function and normal ciliary function in bronchi and prevent the development of chronic infections.
REFERENCES
- Ong T, Ramsey BW. Cystic fibrosis: a review. JAMA 2023;329(21):1859–1871. DOI: 10.1001/jama.2023.8120.
- Boon M, Verleden SE, Bosch B, et al. Morphometric analysis of explant lungs in cystic fibrosis. Am J Respir Crit Care Med 2016;193(5):516–26. DOI: 10.1164/rccm.201507-1281OC.
- Lammertyn EJ, Vandermeulen E, Bellon H, et al. End-stage cystic fibrosis lung disease is characterised by a diverse inflammatory pattern: an immunohistochemical analysis. Respir Res 2017;18(1):10. DOI: 10.1186/s12931-016-0489-2.
- Einarsson GG, Vanaudenaerde BM, Spence CD, et al. Microbial community composition in explanted cystic fibrosis and control donor Lungs. Front Cell Infect Microbiol 2021;11:764585. DOI: 10.3389/fcimb.2021.764585.
- Malhotra S, Hayes D, Wozniak DJ. Mucoid Pseudomonas aeruginosa and regional inflammation in the cystic fibrosis lung. J Cyst Fibros 2019;18(6):796–803. DOI: 10.1016/j.jcf.2019.04.009.
- Qin S, Xiao W, Zhou C, et al. Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct Target Ther 2022;7(1):199. DOI: 10.1038/s41392-022-01056-1.
- Maurice NM, Bedi B, Sadikot RT. Pseudomonas aeruginosa biofilms: host response and clinical implications in lung infections. Am J Respir Cell Mol Biol 2018;58(4):428–439. DOI: 10.1165/rcmb.2017-0321TR.
- Høiby N, Ciofu O, Johansen HK, et al. The clinical impact of bacterial biofilms. Int J Oral Sci 2011;3(2):55–65. DOI: 10.4248/IJOS11026.
- Høiby N, Ciofu O, Bjarnsholt T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol 2010;5(11):1663–74. DOI: 10.2217/fmb.10.125.
- Høiby N, Bjarnsholt T, Moser C, et al. Diagnosis of biofilm infections in cystic fibrosis patients. APMIS 2017;125(4):339–343. DOI: 10.1111/apm.12689.
- Høiby N. A short history of microbial biofilms and biofilm infections. APMIS 2017;125(4):272–275. DOI: 10.1111/apm.12686.
- Lichtenberg M, Coenye T, Parsek MR, et al. What’s in a name? Characteristics of clinical biofilms. FEMS Microbiol Rev 2023. DOI: 10.1093/femsre/fuad050.
- Durda-Masny M, Goz´dzik-Spychalska J, John A, et al. The determinants of survival among adults with cystic fibrosis-a cohort study. J Physiol Anthropol 2021;40(1):19. DOI: 10.1186/s40101-021-00269-7.
- Høiby N. Understanding bacterial biofilms in patients with cystic fibrosis: current and innovative approaches to potential therapies. J Cyst Fibros 2002;1(4):249–54. DOI: 10.1016/s1569-1993(02)00104-2.
- Martin I, Waters V, Grasemann H. Approaches to targeting bacterial biofilms in cystic fibrosis airways. Int J Mol Sci 2021;22(4). DOI: 10.3390/ijms22042155.
Article citation: Nugent K. Cystic fibrosis and pulmonary biofilms. The Southwest Respiratory and Critical Care Chronicles 2023;11(49):19–26
From: Department of Internal Medicine, Texas Tech University Health Sciences Center, Lubbock, Texas
Submitted: 9/5/2023
Accepted: 9/21/2023
Conflicts of interest: none
This work is licensed under a Creative Commons
Attribution-ShareAlike 4.0 International License.